
Article #7 of a 7 Part Series
Alzheimer’s disease is a gradual change in the brain that occurs for decades before mental dysfunction is noticed.
Abundant loss of neurons, and a resulting decrease in brain volume proceeds sequentially through specific areas of the brain associated with memory and reasoning. Neuron loss begins at the entorhinal and hippocampus cortex, areas devoted to memory coding, recall and updating.
An estimated 44 million people worldwide lived with dementia in 2014. The number is expected to rise to 70 million by 2030. Alzheimer’s disease accounts for 60% to 80% of all cases of dementia. By 2050 the projected cost of caring for individuals with Alzheimer’s disease could reach $1.2 trillion in the United States alone.
Prodromal Alzheimer’s disease is a very early form of the disease where the person is still functional.
Therapies for Alzheimer’s disease since 2006 failed at a rate of 95% across all three phases of clinical testing. So why is Alzheimer’s resistant to treatment?
What clues are there from normal functioning of the brain that may help to dissect where it all goes wrong in Alzheimer’s disease?
Alzheimer’s Brain
The Alzheimer’s brain shows gross changes in its architecture. Alzheimer’s disease is differentiated from other forms of dementia by postmortem examination of the brain. When dementia is caused by Alzheimer’s disease, there is gross shrinkage in the hippocampus and areas of the temporal lobe, parietal lobe and frontal lobe. These brain areas manage language, memory, emotion and the ability to reason.
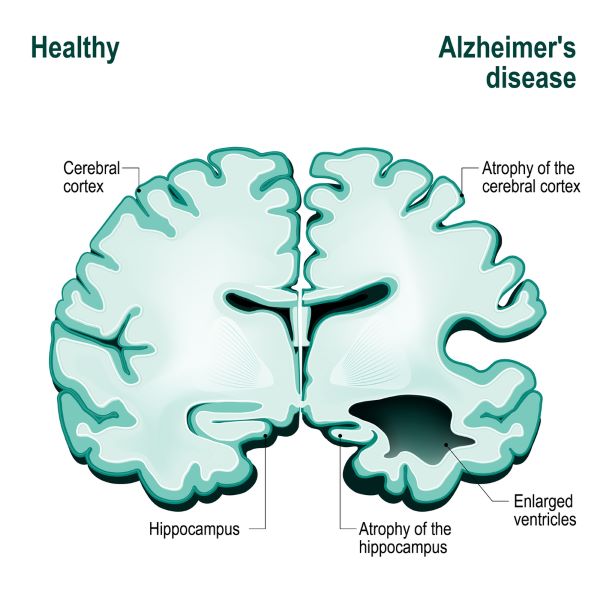
Illustration of what Alzheimer’s does to the human brain, Designua/Shutterstock.com
Postmortem histology of brain tissue detects signs of damage to surviving neurons. Large deposits of a substance called amyloid-beta surrounds damaged neurons. Neuron dendrites are reduced in size and number, and dendrite spines are greatly diminished. The most obvious internal neuron damage is the presence of a fibrous deposit described as tangled tau.
For many years now tangled tau and amyloid beta deposits were thought to be the cause of Alzheimer’s disease. Failure of drug treatments targeting these molecules have cast doubt on that hypothesis.
Amyloid-beta is a normal molecule found at neuron synapses. It is thought to play a role in the effectiveness of neurotransmitter signaling. As the brain ages, there appears to be either an over production of amyloid-beta or the brain loses its ability to clear it, or maybe both.
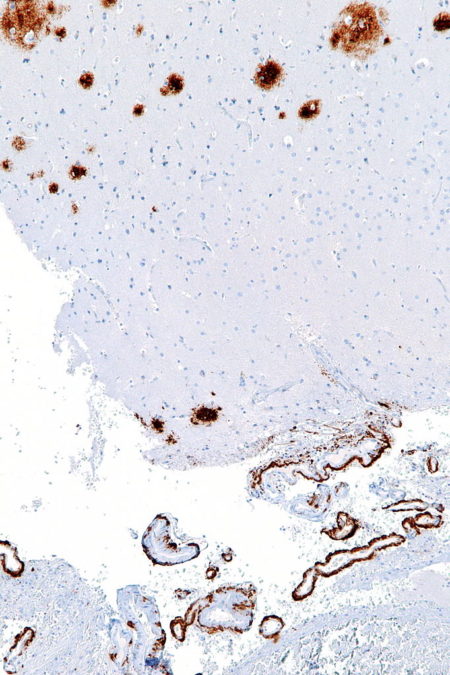
Amyloid-beta Deposits in Cerebral Cortex and Around Blood Vessel, Nephron/Wikimedia Commons
As amyloid-beta concentration rises near neuron synapses it binds to itself creating a multi-stranded ridged structure. Large ridged structures of amyloid-beta shown in this histology section of the human brain are toxic at neuron synapses.
Tau is a soluble protein within healthy neurons. It provides for adequate spacing between the track-like assemblies for moving material from place to place within neurons.
Tau protein lacks the amino acid sequence necessary for a protein’s passage through cell membranes. However, soluble tau is found within synaptic clefts of Alzheimer’s brain, and it is as toxic there as the rigid amyloid-beta.
Reactive Astrocytes
When brain tissue sustains an injury, both protoplasmic and fibrous astrocytes change their character and become reactive astrocytes. Astrocytes are glial cells that perform much of the housekeeping functions within the brain. They are also responsible for repairing the brain when trauma occurs.
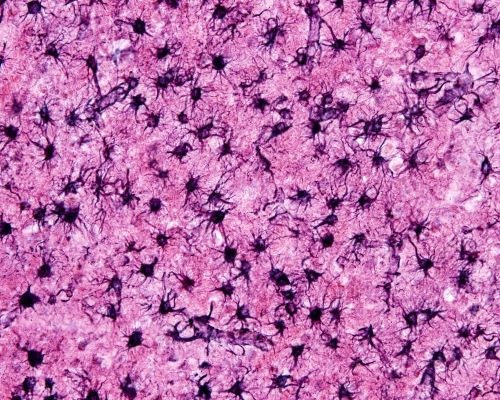
Photomicrograph of protoplasmic astrocytes tiling brain tissue, Jose Luis Calvo/Shutterstock.com
In less severe forms of injury, reactive astrocytes migrate to the site, adjust and repair local brain structure without scar formation. When damage is severe, reactive astrocytes form scars to wall off the damage.
A large number of reactive astrocytes are present in postmortem brain of those suffering Alzheimer’s disease. They are found surrounding amyloid beta deposits.
The shift from an interspersed population of supportive protoplasmic and fibrous astrocytes to a population where reactive astrocytes predominate begins in the initial stages of Alzheimer’s disease.
Careful studies suggest the total population of brain astrocytes does not change during disease progression. Rather the percent of the population functioning as protoplasmic and fibrous astrocytes decreases while the percent functioning as reactive astrocytes increases.
Amoeboid Microglia
Another type of brain cell, microglia normally surveys and monitors the brain for small damage that it can repair, and it removes neuron structures no longer useful like excess dendrites. However, glia is also the resident immune system of the brain and is activated to an amoeboid macrophage-like cell to remove infection.
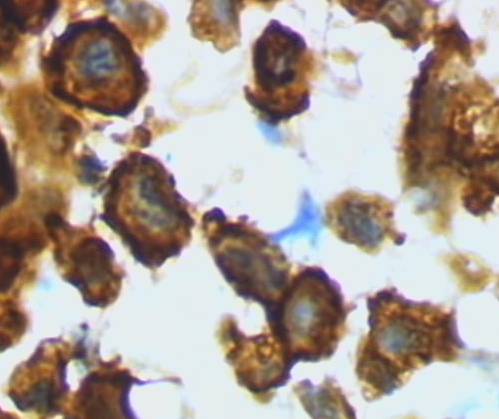
Activated microglia are brain macrophages, Grzegarz Wicker/Wikimedia Commons
Alzheimer’s brain has a large population of the activated, amoeboid macrophage-like form of microglia. Because activated microglia also responds to deposits of multi-stranded amyloid-beta encased in reactive astrocytes as if it is a foreign intruder, Alzheimer’s is often described as an inflammatory disease.
Inflammation in brain tissue is different than Classic Inflammation observed elsewhere in the body. Classic Inflammation includes tissue swelling and heat due to opening of blood vessels to water and white blood cells.
Classical inflammation does not occur in the brain unless the blood brain barrier is broken. The blood brain barrier of deceased Alzheimer’s patients usually shows no functional loss beyond that normally expected because of age.
Development of Alzheimer’s Disease
Development of Alzheimer’s disease has a long pre-symptomatic period. Abnormal amounts of soluble, single-strand, amyloid-beta and soluble tau in cerebrospinal fluid precede mild cognitive decline of Alzheimer’s disease by several years. Amyloid-beta decreases and tau increases from normal amounts during this time.
The decrease in amyloid-beta in cerebrospinal fluid is thought to reflect a reduction in its normal rate of elimination from the brain. Because tau’s normal work is thought to be inside cells, its elevated presence in cerebrospinal fluid suggests there is some abnormal regulation of tau in pre-symptomatic Alzheimer’s disease.
Below normal consumption of glucose in the brain is another characteristic of pre-symptomatic Alzheimer’s disease. People at risk because of variations in their genes, and who develop the disease, sometimes exhibit a decline in their brain’s consumption of glucose 20-30 years prior to symptoms.
Yet, not every person with predisposing genes develops Alzheimer’s disease. And, only about 50% of people with Alzheimer’s disease have genes that are identified as high-risk variants.
PET Imaging
Positron emission tomography (PET imaging), an imaging technique for pre-symptomatic Alzheimer’s disease, is increasingly used even though it is an invasive method. PET requires injection of radioactive molecules that accumulate in the brain.
A radioactive tracer is incorporated into a biologic molecule capable of crossing the blood brain barrier. In the brain, the radioactive molecule emits a signal that can be measured after binding to a brain substance, or after it is modified by a brain enzyme.
The biologic molecules chosen for Alzheimer’s studies include fluorodeoxyglucose (FDG), an analog of glucose, and Pittsburg compound B (PiB), a molecule that binds to amyloid-beta plaques.
With FDG, Alzheimer’s investigators look for brain areas with decreased utilization of glucose. Decreased consumption of glucose is interpreted as decreased neuron electrical signaling.
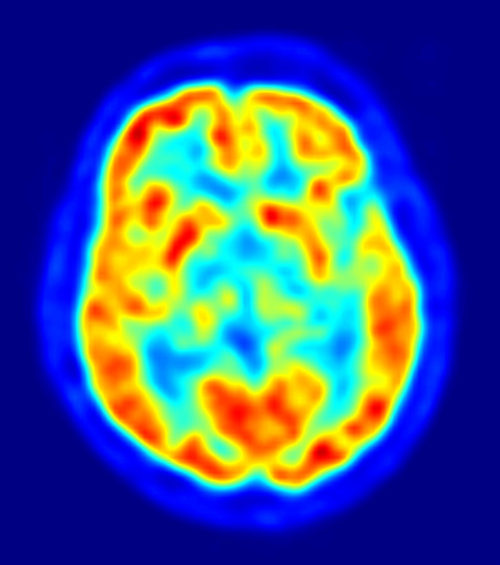
PET Brain Image overlaying MRI Image, Red = High FDG Accumulation, Blue Areas = Low to No FDG, Jens Maus/Wikimedia Commons
PET data can be overlaid on MRI structural data as in this figure for a precise location of areas with low glucose metabolism.
Functional MRI & MEG Studies
Functional magnetic resonance imaging (fMRI), like PET, uses an indirect measurement for evaluating neuron activity. fMRI, a non-invasive method, measures flow of oxygenated hemoglobin into small areas of the brain.
Highly active neurons require a great deal of hemoglobin-carried oxygen to extract energy from glucose molecules.
Also like PET, fMRI data is superimposed on MRI structural data for location of the detected alteration in blood flow. The bright orange and yellow in this image marks the presence of highly active neurons.
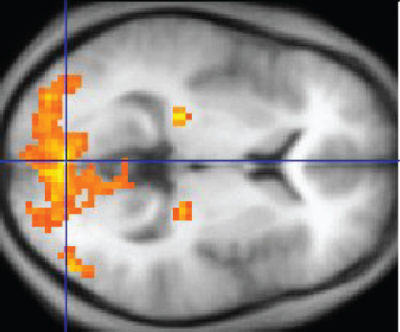
fMRI Data Superimposed on MRI, OpenStax College/Wikimedia Commons
In contrast to PET and fMRI, magnetoencephalography (MEG), another non-invasive method, directly measures actual neuron electrical activity. Although too expensive for most research, MEG is being used to some extent for studies of Alzheimer’s disease.
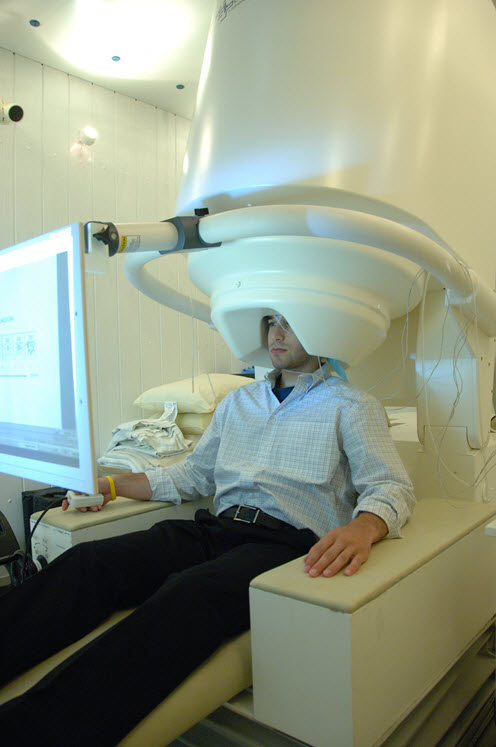
Person undergoing MEG, Public Domain Image/Wikimedia Commons
One theory based on fMRI and MEG data proposes that multiple functional rearrangements occur during the pre-symptomatic phase of Alzheimer’s disease. Neuron re-routing of information is estimated to happen as early as 10 years before symptoms appear.
Cause of Alzheimer’s Disease
Nun Study of Dementia
A feature lacking in studies of Alzheimer’s disease up until the publication of the Nun Study in 1997 was a control group of brains from people who lived long and did not develop dementia. A similar but larger ongoing study, the Religious Orders Study is now accumulating and analyzing over 20 years of clinical and brain histology data on more than 1,000 participants.
Nuns, priests and brothers from 31 Catholic orders in the United States are participating in the Religious Orders Study. The study is funded by the National Institute of Health on Aging at Rush University Medical Center in Chicago.
A surprising outcome is that amyloid-beta plaques and tau tangles in the brain do not always result in neuron depletion and dementia in all individuals. There is increasing evidence that there is a dissociation between Alzheimer’s dementia and the chief biomarkers of the disease, at least in some individuals.
Among participants without cognitive impairment at death, 12% had brains with an equivalent burden of amyloid-beta plaque and tau tangles as brains of those experiencing Alzheimer’s dementia. However, in these unusual cases, there was a preservation of neurons, synaptic structures and axon geometry and a lack of reactive astrocytes and microglia.
A more in depth discussion of data from the Religious Orders Study and Nun Study can be found in a free paper at PubMed, “Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology”, Perez-Nievas BG et al., 2013, Brain 136:2510-2526.
Amyloid beta & Tau
Amyloid beta and tau until recently were thought to cause Alzheimer’s disease. It was believed for decades that removal of amyloid-beta plaque would halt disease progression for patients assumed to be experiencing Alzheimer’s dementia. Clinical trials involved delivery of antibody drugs that were able to cross the blood brain barrier.
The antibody was expected to assist the brain’s microglia destroy the amyloid-beta deposits. Clinical trial data confirmed that antibody did indeed pass through the blood brain barrier and clear amyloid-beta plaques.
Unfortunately, treating patients with mild to moderate dementia, who also had genes predisposing them to Alzheimer’s disease, with antibody against amyloid-beta failed to delay progress of the disease.
Lack of positive results in clinical trials targeting amyloid-beta plaque led some investigators to question whether its presence in Alzheimer’s is a cause or symptom of the disease.
Several investigations are now proceeding to test whether removal of toxic extracellular soluble tau from the brain may provide better results.
Rogue Microglia?
A suspect in the development of Alzheimer’s disease is microglia gone rogue. As Alzheimer’s disease progresses, astrocyte and microglia reactivity increase linearly around plaque and tangles. Of note, in Alzheimer’s disease all dead neurons are missing from the brain. Dead and dying neurons are not identified in postmortem tissue.
In healthy brain, the surveying form of microglia monitors the well-being of synapses. If a neuron is damaged, surveying microglia make an attempt to repair it by secreting growth factors. If the neuron is damaged beyond repair, surveying microglia changes to a macrophage-like form and kills the neuron and removes the debris.
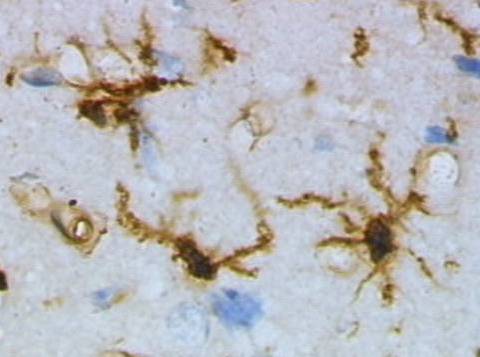
Brain Tissue Stained to Display Microglia, Grzegarz Wicker/Wikimedia Commons
It is unknown in Alzheimer’s disease whether surveying microglia fails to repair damage to neurons caused by some unknown factor, or if surveying micoglia plays a role in initiating neuron death before assuming its macrophage-like form and disposing of the remains.
Metabolic Hypothesis
Neuron’s share glucose metabolic pathways with astrocyte glia. A decrease in glucose metabolism revealed by PET with FDG aligns with the onset of brain atrophy detected by MRI. MRI data indicate the entorhinal cortex shows significant volume change 8-10 years before onset of symptoms. The hippocampus shows atrophy beginning 2-4 years prior to symptoms.
Neurons depend almost exclusively on glucose for energy. Metabolic reliance of neurons on astrocyte molecules for efficient energy production is modeled by scientists as a complex process. This may be an important relationship if the metabolism hypothesis is correct.
The metabolism hypothesis states that a decrease in neuron glucose utilization is the initial step in development of Alzheimer’s disease. If true, the metabolic defect could be an astrocyte defect, or a neuron defect, or both.
Interestingly, cognitively normal subjects in the Religious Orders Study and Nun Study with a high load of amyloid-beta and tau also had a normal appearing brain astrocyte population, unlike Alzheimer’s individuals.
Cholesterol Hypothesis
Cholesterol is among the lipids that cannot cross the blood brain barrier. There is no mixing of brain cholesterol with cholesterol in the blood. All cholesterol used by the brain is made in the brain primarily by astrocytes and oligodendrocytes.
Cholesterol is critical to neuron synapse and dendrite formation, axon elongation, and it is a major component of myelin and neurotransmitter vesicles. For transport within the brain, cholesterol is wrapped in a protein name ApoE. ApoE is produced by astrocytes and microglia and is taken up by brain cells using a receptor named LRP1.
The same LRP1 receptor, on astrocytes at the blood brain barrier, is also the major mechanism for removal of soluble amyloid beta from the brain. Some data indicate single-strand amyloid-beta competes with ApoE for use of the LRP1 receptor.
ApoE is produced normally as 3 inheritable alleles, ApoE2, ApoE3 and ApoE4. The ApoE4 variant is a major risk factor for late onset of Alzheimer’s disease. At conception, each person receives two alleles of each gene, one from father and one from mother. Approximately 50% of people with late-stage Alzheimer’s disease have at least one ApoE4 allele.
One hypothesis is that ApoE4 binding to LRP1 blocks elimination of amyloid-beta from brain, encouraging plaque formation. Research into ways to make ApoE4 work more like ApoE3 in the brain is ongoing.
Head Trauma & Dementia
It is too early to come to conclusions about the connection between multiple head trauma and subsequent dementia. Dementia studies require a substantial number of participants, a willing control group and at least 20 years of data, as was learned with study of Alzheimer’s disease.
There may, however, be hints in the way the brain handles trauma for an appropriate approach to such studies. Neurons respond to areas of injury in the brain by adjusting their circuits in undamaged sections to by-pass failure of the damaged tissue.
Isolation of the damaged tissue followed by repair or elimination of damaged tissue is left to astrocytes, oligodendrocytes and microglia.
Astrocytes respond to all types of central nervous system injury. A response known as reactive astrogliosis is a hallmark of structural damage in the brain. It is a graded response.
With a mild to moderate amount of damage there is no extensive reorganization of tissue structure by astrogliosis. With resolution of the injury, astrocytes return to their normal state.
For a highly destructive insult like torn tissue, astrogliosis produces lasting reorganization of the tissue and scar tissue along the edges of the tissue destruction. Astrocytes interact with oligodendrocytes and microglia and cells of the meninges to deposit dense collagen.
The deposited material contains molecular signals to inhibit neuron reorganization within the damaged area. In this form of astrogliosis, structural changes in the brain tissue last long after the injured section is enclosed.
Between the response to non-concussion injury and torn tissue there are many types of graded astrogliosis. This makes discovering an appropriate biomarker for the connection between repeated head trauma and late-onset dementia challenging.
But, studying astrocyte responses to repeated head trauma may be a place to begin learning the connection between repeated head injury and dementia later in life.
I hope that I have convinced you during this Virtual Tour Inside the Closed World of the Brain that answers for modifying brain diseases will be found in research leading to a better understanding of how healthy human brain cells connect with each other, share the work and efficiently disengage in a timely fashion.
If you have questions or comments, please email me at DrReece@MedicalScienceNavigator.com.
 Margaret Thompson Reece PhD, physiologist, former Senior Scientist and Laboratory Director at academic medical centers in California, New York and Massachusetts is now Manager at Reece Biomedical Consulting LLC.
Margaret Thompson Reece PhD, physiologist, former Senior Scientist and Laboratory Director at academic medical centers in California, New York and Massachusetts is now Manager at Reece Biomedical Consulting LLC.
She taught physiology for over 30 years to undergraduate and graduate students, at two- and four-year colleges, in the classroom and in the research laboratory. Her books “Physiology: Custom-Designed Chemistry”, “Inside the Closed World of the Brain”, and her online course “30-Day Challenge: Craft Your Plan for Learning Physiology”, and “Busy Student’s Anatomy & Physiology Study Journal” are created for those planning a career in healthcare. More about her books is available at https://www.amazon.com/author/margaretreece. You may contact Dr. Reece at DrReece@MedicalScienceNavigator.com, or on LinkedIn
Reece Biomedical Consulting LLC Privacy Policy: Click HERE
Featured header image: ©Moonlight Photo Studio, via Shutterstock.com
![]()